We use an atomistic approach based on forcefields to simulate how hydrocarbons diffuse through the microporous structure of zeolites. I employ the general purpose DLPOLY code which I recommend for it is robust, portable, easily modifyable, and quick in serial and parallel machines.
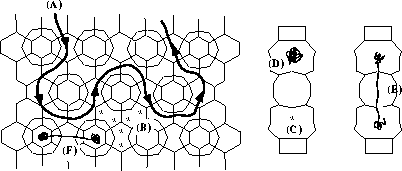
The MCM-22 structure showing the general
features of molecules diffusing through it. (A) A molecule diffuses through the
10 MR sinusoidal system. (B) Potential energy minima in the sinusoidal system.
(C) Potential minima in the large cavities. (D) Intracage motion in the large
cavities around a minimum. (E) Intracage motion around the two minima in the
large cavity. (F) Intercage diffusion through the large cavities.
The simulations allow us to calculate diffusion properties, very especially
the magnitude called "diffusivity", and also to visualise trajectories of
the individual molecules through the intricated microporous zeolite. We can
see whether the molecule/s remain in a restricted part of the solid, how
do they diffuse in the different channels and cavities, the time spent in
each channel, the individual diffusion coefficients through each channel,
the interactions between molecules in each channel or in the global structure,
the activation energies or energy profiles for each molecule, and the radial
distribution funtions among other magnitudes. So we can get a clear picture
of the physics of the process. Then, we establish comparisons of a given
sorbate in different zeolites, or different sorbates in one zeolite, or we
can study the effect of loading, or the effect of temperature, or we can
compare diffusion in single component and mixed component systems.
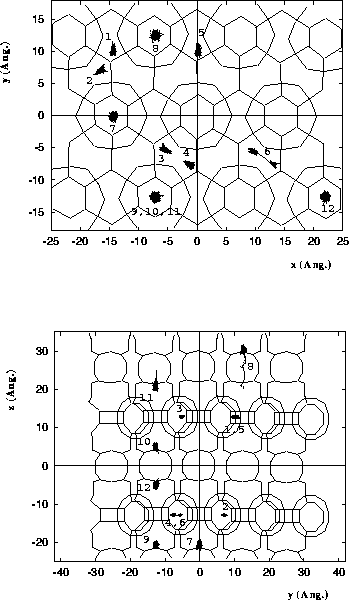
Diffusion of 2-methyl-hexane in MCM-22.
Molecules 1-6 are located in the
10 MR channels, and molecules 7-12 are
located in the 12 MR supercage systems.
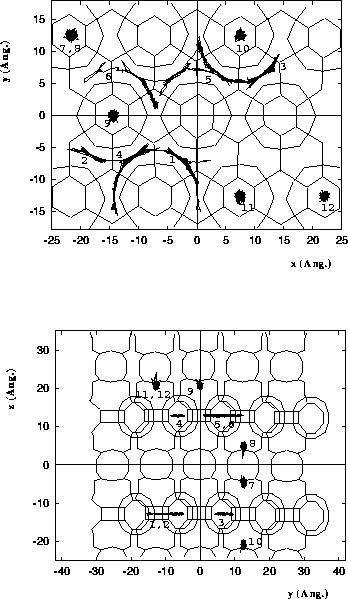
Diffusion of n-heptane in MCM-22.
Molecules 1-6 are located in the
10 MR channels, and molecules 7-12 are
located in the 12 MR supercage systems.